Original Course Description CPANG19
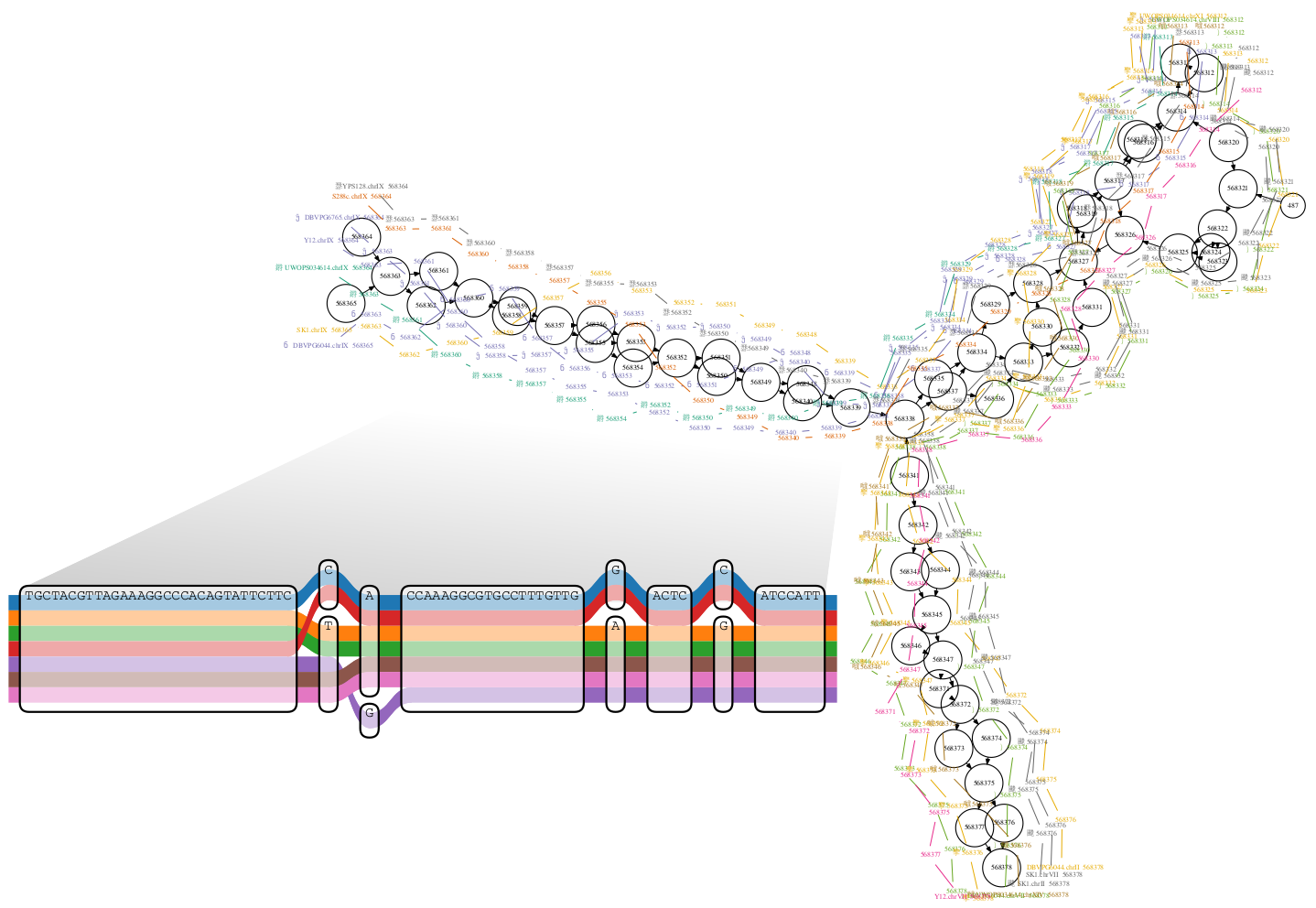
Reference genomes have become central to bioinformatics approaches, and form the core of standard analyses using contemporary sequencing data. However, the use of linear reference genomes, which provide the sequence of one representative genome for a species, is increasingly becoming a limitation as the number of sequenced genomes grows. In particular, they tend to bias us away from the observation of variation in the genomes we study. A general solution to this problem is to use a pangenome that incorporates both sequence and variation from many individuals as our reference system. This pangenome is naturally modeled as a graph with annotations, and can provide all the functionality traditionally provided by linear reference genomes. Unlike linear reference genomes, a pangenome readily incorporates both small and large variation, allowing bias-free genotyping at known alleles. In this course we will explore the use of modern bioinformatic tools that allow researchers to use pangenomes as their reference system when engaging in studies of organisms of all types. Such techniques will aid any researcher working on organisms of high genetic diversity or on organisms lacking a high-quality reference genome. This course targets all researchers interested in learning about an exciting paradigm shift in computational genomics.
This Project
In this project, different tools for computational pangenomics are installed and tested, with one focus on the original course materials and another on the bacterial pathogen Pseudomonas aeruginosa.